


- Research
 Evolution of Biophysical Genotype Phenotype maps
Evolution of Biophysical Genotype Phenotype mapsUsing analogies from statistical physics to understand emergent phenomena in evolution
Natural selection acts on phenotypes, yet variation in phenotypes arises from mutations at the genetic level. It is being increasingly recognised that a crucial missing ingredient for a full understanding of evolution, even at a qualitative level, is the role of genotype-phenotype maps, or the mapping from sequence to function. Quantitative and predictive theories of evolution that account for this mapping will be required to understand several pressing problems, including antibiotic resistance, virus evolution, and cancer evolution. A key generic prediction is that small drift-dominated populations adapt to phenotypes that have the most sequences coding for them instead of the most optimal (Khatri, PNAS 2009, Genetics 2016). This means smaller populations undergo more rapid speciation as common ancestors are more likely to be maladapted (Khatri, JTB 2015, Khatri, Genetics, 2015 & Khatri, PLoS Computational Biology 2019). In addition, I have developed a stochastic theory for the evolution of phenotypes within a continuous stochastic dynamics framework (Khatri et al, JTB, 2015); this holds promise of greatly simplified modelling of complex genotype-phenotype maps in the future.
 Virus Evolution
Virus EvolutionDetecting selection in longitudinal deep sequencing of viruses
My work aims to understand the basic evolutionary forces that shape the genomes of pathogens and the stochastic dynamics of variants in a host. A key quantity to calculate is the probability of observing a change in the frequency of a variant in a fixed time interval. This is a difficult mathematical diffusion problem that has eluded accurate solution for changes over short times. The key difficulty is the that the variance of allele frequencies (or diffusion constant) is not the same for all allele frequencies. Fisher's angular transformation gives a variance which is independent of a new angular frequency and so effectively simple Brownian motion. The cost is a non-linear potential which is a manifestation of the effects of genetic drift, and is probably why this transformation has largely gone unnoticed and unexploited, despite Fisher discovering it almost a century ago. To make practical use of the transformation, I developed an heuristic Gaussian-based method that gave simple and accurate solutions including genetic drift, selection and mutation. This work (Khatri, Sci. Rep. 2016) is a key foundational basis for analysing longitudinal deep-sequencing data. More recently, I used the same framework for a new analysis which can distinguish between transmission and intrahost fitness effects in phylogenetic trees (Khatri et al, in preparation) and a new method to calculate the rate of fixation of rare variants (Khatri, bioRxiv123232, 2017).
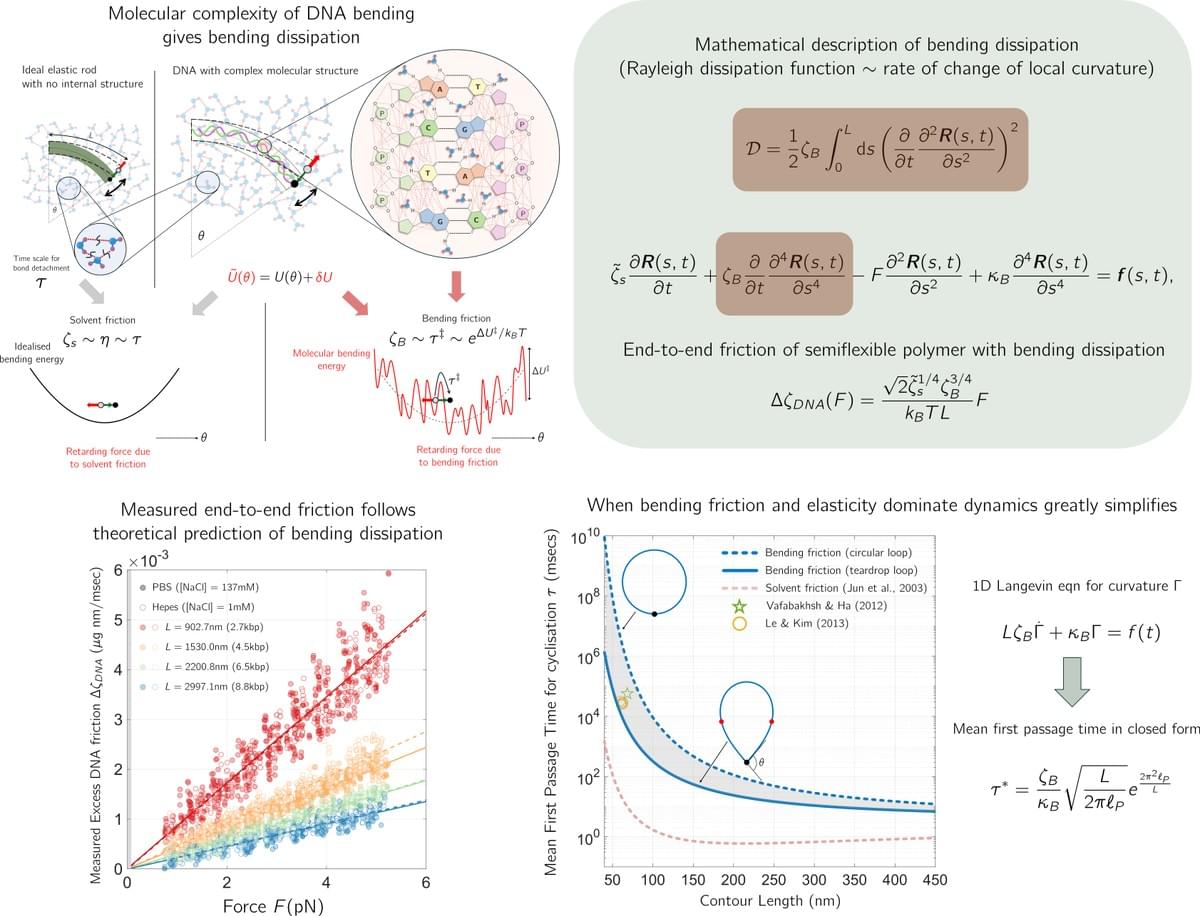
New polymer physics of DNA bending dissipation
A new material constant of DNA
DNA bending and looping is crucial for gene expression, packaging, and chromatin organisation. Yet current theories of DNA dynamics based on the semiflexible polymers underestimate empirically measured DNA loop-closure times by a remarkable 1000-fold. We have lacked a basic understanding of what determines how quickly DNA bends? With experimental biophysicist Maxim Moldtsov (Francis Crick Institute and Department of Physics, UCL), we have discovered that for lengths of DNA between 50 nm and 400 nm it is not solvent friction — as semiflexible polymer theories assume — but a new material constant of DNA, the bending friction, that determines how quickly DNA responds to mechanical forces. We determine this new material constant of DNA — from single molecule stretching experiments using optical tweezers — to be ζB = 241±17 μg/msec and strikingly independent of changing salt concentration (unlike the bending elasticity). This reveals a new picture of DNA mechanics at these length scales: rather than an ideal elastic rod, DNA is a viscoelastic polymer with a locally complex energy landscape for bending with dramatically slower dynamics. This is a fundamental discovery on par with establishing DNA’s elastic properties, which are well-described by the Worm-Like Chain model. While the bending elasticity has been profoundly influential, it is likely that bending friction and the Dissipative WLC (DWLC) model will be even more so: molecular biology is inherently out-of-equilibrium. This work has established a new foundation for DNA dynamics: any biological process involving DNA looping — from the action of molecular machines to chromosome organisation —
must account for this friction.
Contact


Email
bhavin.khatri@physics.org

Research Fellow in Statistical Genomics
Imperial College London
Department of Life Sciences
Silwood Park
&
Visiting Scientist
The Francis Crick Institute
London



